a freeware tool for 2D depiction of molecular structures
Availability
mol2ps is freely available under the terms of the GNU General Public License (GPL), for a detailed description of this license, please visit http://www.gnu.org/copyleft/gpl.html.Download
Installation
Download the appropriate compressed binary file for your system, uncompress it and rename the resulting executable file intomol2ps
(for Linux, Mac OS X, or FreeBSD), for Windows there is no need to rename the
extracted file, mol2ps.exe. Copy
this program file into a directory which is in your search path (e.g., /usr/local/bin
or C:\UTIL). There are no additional files or
libraries required. For mol2svg, copy or rename/hard-link the mol2ps executable into mol2svg (Windows: mol2svg.exe).Usage (command-line options)
mol2ps can be invoked with the following arguments (version 0.4)mol2ps [options] <filename>
where <filename> is the file containing the
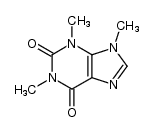
molecular structure
(supported formats: MDL *.mol or *.sdf, Alchemy *.mol, Sybyl
*.mol2)
if <filename> is "-" (without quotes), the program reads
from standard input
valid options are:
-R (reaction mode, for MDL rxn and rdf files)
--font=<Helvetica|Times>, default: Helvetica
--fontsize=<any number in points>, default: 14
--fontsizesmall=<any number in points>, default: 9 (for
subscripts)
--linewidth=<n.n>, default: 1.0 (linewidth in points; use
1 decimal)
--rotate=<auto|auto3Donly|n,n,n>, default: auto (n,n,n
specifies the
angles to rotate the molecule around the X, Y, and Z
axis (in degrees)
--autoscale=<on|off>, default: on (scales the molecule to
fit the natural
C-C bond length)
--striphydrogen=<on|off>, default: on (strips all explicit
H atoms)
--hydrogenonhetero=<on|off>, default: on (adds H to all
hetero atoms)
--hydrogenonmethyl=<on|off>, default: on (adds H to all
methyl C atoms)
--hydrogenonstereo=<on|off>, default: on (shows H if bond
is "up" or "down")
--showmolname=<on|off>, default: off (prints name above
the structure)
--atomnumbers=<on|off>, default: off (prints atom numbers)
--bondnumbers=<on|off>, default: off (prints bond numbers)
--sgroups=<on|off>, default: on (uses Sgroup abbreviations if present)
--showmaps=<on|off>, default: off (prints atom-atom mapping numbers)
--color=</path/to/color.conf>, default: no colors for
atoms labels
--bgcolor=<white|gray|n,n,n> where n,n,n are the RGB values (0-255)
--scaling=<n.n>, default: 1.0 (any scaling factor from 0.1 to 10.0)
--output=<ps|eps|svg>, default depends on prog name (mol2ps, mol2eps, mol2svg)
mol2ps mymolecule.mol > mymolecule.psFinetuning
The program attempts by default to orient the molecule in a suitable way if it contains a ring structure. Especially with 3D molfiles, this is quite useful, but sometimes can give unsatisfactory results. Please inspect the generated Postscript file with a text editor: in the file header, the applied angles of rotation around the X, Y, and Z axis are noted as Postscript comments (lines starting with a % sign). You can take these values as starting points for some manual optimization, using the command-line argument "--rotate=" (see above),
for example --rotate=10,-23,75 (which would rotate the
structure by 10° around X, -23° around Y, and 75° around Z).Useful Perl scripts for batch processing
If you want to convert an MDL molfile or an entire SD file directly into 2D bitmap pictures in PNG format, take a look at the Perl script mol2png.pl. For conversion of an already existing Postscript file into a PNG image file, the script ps2png.pl is available. These scripts use Ghostscript not only for rastering, but also to determine suitable dimensions of the output bitmap file (using the "bbox" device of Ghostscript). Of course, the resulting PNG bitmap files can be converted into any other bitmap format (such as GIF) by almost any graphics conversion utility program.