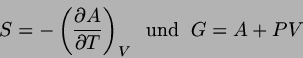Wir bringen nun wieder Systeme miteinander in
thermischen
Kontakt; dabei soll aber eines der beiden Systeme sehr viel
mehr Freiheitsgrade aufweisen als das andere. Es soll also gelten
(4.19)
Abbildung 4.2:
System in Kontakt mit einem Energiespeicher:
kanonische Gesamtheit
Das größere System, mit
Freiheitsgraden, wird als
,,Wärmebad`` bezeichnet. Die Energie
dieses
Wärmebades ist ,,fast immer`` viel größer als die des
kleineren Systems, und seine
Entropie kann daher um
entwickelt werden:
(4.20)
wobei die Temperatur des Wärmebades ist. Damit wird aber
die Zellenanzahl, die
das größere System in seinem Phasenraum einnimmt, wenn System in einem
Zustand der Energie ist,
(4.21)
Je größer aber
ist, umso wahrscheinlicher ist es,
daß das System einen Mikrozustand der Energie aufweist. Dies
läßt sich mit Hilfe der ,,kanonischen Phasenraumdichte``
so ausdrücken:
(4.22)
wo ein Normierungsfaktor ist und
(4.23)
die Dichte der Mikrozustände in jenem Bereich des Phasenraums von
System bezeichnet, der zu der Energie gehört.
Zum besseren Verständnis von 4.22 erinnern wir uns daran,
daß im mikrokanonischen Ensemble nur jene Zustände des Systems
betrachtet wurden, für die die Energie im Intervall
lag. Im kanonischen
Ensemble sind alle Energiewerte zugelassen, die Dichte
der Zustandspunkte variiert aber wie
.
Gleichung 4.22 darf nicht so verstanden werden, daß die
wahrscheinlichste Energie des kleinen Systems gleich
wäre. Die Dichte der Zustände im Phasenraum von System
nimmt zwar mit anwachsendem ab, das zu gehörige
Volumen nimmt aber zu - und zwar mit
, also sehr stark. Das Produkt dieser beiden
Faktoren, also das statistische Gewicht des Phasenraumbereichs, der zu
gehört, weist daher ein Maximum bei einer Energie
auf.
Ein nun schon wohlbekanntes Beispiel zur Illustration des eben
Gesagten bietet die Maxwell-Boltzmann-Verteilung:
ist ja nichts anderes als die Wahrscheinlichkeitsdichte für die
(kinetische) Energie eines Teilsystems, das aus nur einem einzigen
Teilchen besteht, und das mit einem Wärmebad aus Teilchen in Kontakt
steht.
Im allgemeinen, nämlich für große Teilchenzahlen, ist das
Maximum der Energieverteilung so scharf,
daß die wahrscheinlichste Energie mit der mittleren Energie zusammenfällt:
(4.24)
Auch Systeme, die untereinander Energie austauschen können, weisen
also praktisch immer, oder jedenfalls mit überwältigender Wahrscheinlichkeit,
eine bestimmte Energie auf. Diese Tatsache erlaubt es uns aber,
Mittelungen physikalischer Größen wahlweise - je nach mathematischer
Bequemlichkeit - im mikrokanonischen oder im kanonischen
Ensemble durchzuführen; man spricht in diesem Zusammenhang von der
,,Äquivalenz der Gesamtheiten``.
Wir haben die Eigenschaften des kanonischen Ensembles mit Hilfe einer
Taylorentwicklung der Entropie hergeleitet. Die von Gibbs ursprünglich
angegebene Herleitung sieht anders aus. Gibbs verallgemeinerte das
von Boltzmann angegebene ,,Verfahren der wahrscheinlichsten
Verteilung`` auf ein Ensemble von makroskopisch gleichartigen
Systemen, die miteinander in thermischem Kontakt stehen.
Gibbs betrachtete gleichartige Systeme (
) mit je
Teilchen. Die Summe der Systemenergien sollte dabei einen
vorgegebenen Wert
einhalten; dabei sollte
aber ein ungehinderter Austausch von Energie zwischen den Systemen
zugelassen sein. Unter diesen einfachen Voraussetzungen bestimmte er
die Wahrscheinlichkeitsdichte dafür, ein System in der Umgebung eines
Mikrozustandes zu finden, dem eine Energie
im Intervall
zukommt. Diese Wahrscheinlichkeitsdichte nimmt mit wachsender Energie
wie
ab. Da aber das Volumen
der Energieschale mit wachsender Energie stark anwächst, lautet das Ergebnis,
daß die meisten Systeme eine Energie um
aufweisen werden. Die wichtige Äquivalenz von kanonischem und
mikrokanonischem Ensemble ist also auch auf diesem Wege belegbar.
THERMODYNAMIK IM KANONISCHEN ENSEMBLE
Die Größe
(4.25)
wird als kanonische Zustandssumme bezeichnet. Sie spielt
zunächst
einmal die Rolle eines Normierungsfaktors bei der Berechnung von
Mittelwerten über das kanonische Ensemble. So ist
beispielsweise
die innere Energie gegeben durch
(4.26)
Die große praktische Bedeutung der Zustandssumme besteht aber darin,
daß sie eng mit der Helmholtzschen freien Energie zusammenhängt,
die ja eine zentrale Größe der Thermodynamik ist. Der Zusammenhang
lautet
(4.27)
wo
. Zum Beweis für diese Behauptung differenzieren
wir die Identität
(4.28)
nach ; so erhalten wir
(4.29)
oder
(4.30)
Dies ist, mit
,
identisch mit der wohlbekannten thermodynamischen Relation .
Alle übrigen thermodynamischen Größen können nun aus
berechnet werden. So ist z. B. der Druck gegeben durch
(4.31)
und Entropie bzw. Gibbssche freie Energie können aus
also die wohlbekannte Zustandsgleichung des idealen Gases. Ebenso erhält
man aus
die Entropie im Einklang mit Gl.
3.39.
Beispiel 2: Die freie Energie von einem Mol eines idealen
Gases bei Normalbedingungen hat den Betrag (für eine Molekülmasse
)
(4.37)
Damit ist es uns gelungen, einzig aus der Betrachtung des
Phasenraums von klassischen Massenpunkten die Thermodynamik
des idealen Gases herzuleiten. Aber auch für andere
Modellsysteme, mit je ihren
zugehörigen Phasenräumen, lassen sich entsprechende Beziehungen
für die beobachtbaren thermodynamischen Größen ableiten.
Das Konzept der ,,Zustandssumme`` läßt sich im Rückblick auch
auf das mikrokanonische Ensemble anwenden. Die dort definierte Größe
war ja ebenfalls ein Maß für das Gesamtgewicht des
zugänglichen Phasenraumvolumens. Man könnte sie daher durchaus als
,,mikrokanonische Zustandssumme`` bezeichnen. Und auch in diesem Fall
diente ihr Logarithmus - also die Entropie - als Ausgangspunkt für die
statistische Thermodynamik.
DER GLEICHVERTEILUNGSSATZ
Ohne Beweis sei der folgende wichtige Satz vermerkt:
Beispiel 1:
Beim klassischen idealen Gas ist die Hamiltonfunktion gegeben durch
(4.38)
Jeder der Freiheitsgrade der Translation kommt also in der Form
vor. Der Gleichverteilungssatz besagt nun, daß für jede
Geschwindigkeitskomponente gilt
(4.39)
Beispiel 2:
Jedes klassische Fluid, wie z. B. das Lennard-Jones-Fluid,
hat bei beliebiger
Dichte die Hamiltonfunktion
(4.40)
Jeder der translatorischen Freiheitsgrade enthält
daher im Mittel die
Energie .
Beispiel 3:
Ein System aus eindimensionalen harmonischen Oszillatoren ist durch die
Energiefunktion
(4.41)
gekennzeichnet. Daher gilt
(4.42)
Die gesamte Energie der Oszillatoren beträgt daher .
Die Verallgemeinerung auf 3 Dimensionen ist trivial; sie führt zu dem
Ergebnis
. Für die spezifische Wärme ergibt sich
somit
. Diese Voraussage
stimmt mit experimentellen Ergebnissen für kristalline Festkörper bei
hohen Temperaturen überein. Bei niedrigen Temperaturen - und damit kann,
je nach Substanz, durchaus auch Zimmertemperatur oder mehr gemeint sein -
bricht die klassische Beschreibung zusammen, und wir müssen
quantenmechanische Regeln anwenden, um und andere thermodynamische
Observable richtig vorhersagen zu können (siehe Kapitel 5).
Beispiel 4:
Ein Fluid aus ,,Hantelmolekülen`` mit je translatorischen und
rotatorischen Freiheitsgraden hat die Hamiltonfunktion
(4.43)
wo die Richtungsvektoren der linearen Moleküle, deren
Winkelgeschwindigkeit und das molekulare Trägheitsmoment bezeichnet.
Somit ist
(4.44)
DAS CHEMISCHE POTENTIAL
Wenn zwei Systeme untereinander Energie austauschen können, führt dies zu
einem Ausgleich der Temperatur (siehe Abschnitt 4.1). Die
Energien der beiden Systeme schwanken darnach - meist nur
geringfügig - um je einen Mittelwert.
Lassen wir nun zu, daß auch Teilchen zwischen den beiden Systemen hin-
und herwechseln können. Auch in diesem Fall wird es zu einem Ausgleich
kommen; darnach werden die Teilchenzahlen nur mehr geringfügig
schwanken. Der Teilchenaustausch wird genau dann zum Stillstand kommen,
wenn die freie Energie des kombinierten Systems, sich
nicht mehr verändert. Das ist dann der Fall, wenn
.
Die Größe, die den Gleichgewichtszustand bestimmt, ist somit
gegeben durch
.
Die beiden Systeme tauschen also so lange Teilchen untereinander aus, bis
. (Man vergleiche diese Definition des
chemischen Potentials mit der Definition der Temperatur,
).)
Beispiel: Das chemische Potential für das obige ideale
Gas bei Normalbedingungen hat den Wert
(4.45)
SIMULATION IM KANONISCHEN ENSEMBLE: DIE MONTE CARLO-RECHNUNG
Auch für die korrekte Durchmusterung der Zustände mit gegebenen
(anstelle von ) läßt sich eine
numerische Methode angeben. Da in diesemFall der Boltzmannfaktor das
statistische Gewicht eines Mikrozustandes
angibt,
gilt für den Mittelwert irgendeiner Größe
, daß
(4.46)
Wenn die Größe nur von Teilchenpositionen, nicht aber von
Geschwindigkeiten abhängt (potentielle Energie, Virial, etc.), genügt
sogar ein gewichtetes Integral
über den -dimensionalen konfigurationellen Unterraum
des
vollständigen Phasenraums;
(4.47)
Man ersetzt die Integrale durch Summen und versucht so einen Schätzwert
für
zu ermitteln. Dazu erzeugt man eine lange Folge von
zufällig ausgewählten Zuständen
,
und zwar so, daß die verschiedenen Zustände darin gleich mit denjenigen
relativen Häufigkeiten auftreten, die ihnen gemäß ihren
Boltzmannfaktoren zustehen;
Konfigurationen mit hoher potentieller Energie kommen also seltener vor
als solche mit kleinem . Die gängige Methode zur Herstellung
einer solchen präparierten Zufallsfolge (einer sogenannten
,,Markov-Kette``) bezeichnet man als ,,Metropolis-Verfahren``,
nach ihrem Erfinder Nicholas Metropolis.
Da der Boltzmannfaktor nun schon in den Häufigkeiten berücksichtigt ist,
kann man den Mittelwert ganz einfach gemäß
(4.48)
ausrechnen. Eine ausführliche Beschreibung der Monte
Carlo-Methode ist in [Vesely 1978] zu finden.
Es gibt übrigens auch eine Art der Molekulardynamikrechnung,
die zur Simulation in der kanonischen Gesamtheit verwendet
werden kann. Normalerweise wird bei MD-Rechnungen ja die
gesamte Systemenergie, also
,
konstant gehalten, was einer Durchmusterung der mikrokanonischen
Gesamtheit entspricht. Führt man aber eine Art von numerischem
Thermostaten ein, der bei jedem Zeitschritt die
Teilchengeschwindigkeiten (und damit ) geeignet
adjustiert, dann kann man entweder erzwingen
(isokinetische MD-Simulation) oder
(isotherme MD).
F. J. Vesely / StatPhys Tutorial, Deutsch, 2001-2003
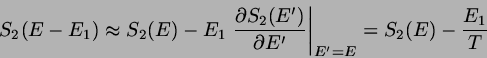
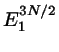 , also sehr stark. Das Produkt dieser beiden
Faktoren, also das statistische Gewicht des Phasenraumbereichs, der zu
, also sehr stark. Das Produkt dieser beiden
Faktoren, also das statistische Gewicht des Phasenraumbereichs, der zu
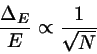
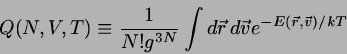
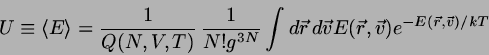
![\begin{displaymath}
\frac{1}{N! g^{3N}} \int d\vec{r} d \vec{v}
\; e^{\beta \left[A(N,V,T)-E(\vec{r},\vec{v} ) \right]}
=1
\end{displaymath}](img814.png)
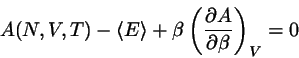
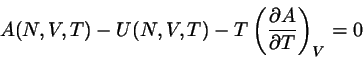
![\begin{displaymath}
Q(N,V,T) = \frac{m^{3N}}{N!h^{3N}}
\int d\vec{r}d\vec{v} \e...
...T}\sum \vert\vec{v}_{i}\vert^{2}]}
= \frac{1}{N!h^{3N}} q^{N}
\end{displaymath}](img821.png)
![\begin{displaymath}
q \equiv \int d\vec{r}d\vec{v} \exp{[-\frac{m}{2kT} \vert\vec{v}\vert^{2}]}
= \left( \frac{2 \pi k T}{m}\right)^{3/2} V
\end{displaymath}](img822.png)
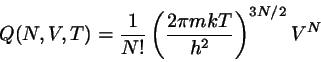
und 4.31
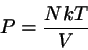
die Entropie im Einklang mit Gl. 3.39.
)
![\fbox{
\parbox[c]{420pt}{
Wenn in der {Energiefunktion} (Hamiltonfunktion) eine ...
...\uml {a}lt
der zugeh\uml {o}rige Freiheitsgrad im Mittel die Energie $kT/2$.
}
}](img829.png)
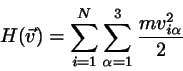
Freiheitsgrade der Translation kommt also in der Form
vor. Der Gleichverteilungssatz besagt nun, daß für jede Geschwindigkeitskomponente gilt
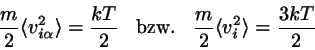
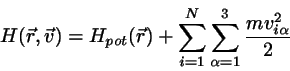
.
eindimensionalen harmonischen Oszillatoren ist durch die Energiefunktion
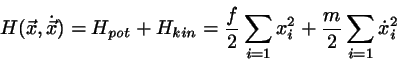
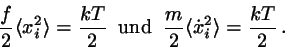
. Die Verallgemeinerung auf 3 Dimensionen ist trivial; sie führt zu dem Ergebnis
. Für die spezifische Wärme ergibt sich somit
. Diese Voraussage stimmt mit experimentellen Ergebnissen für kristalline Festkörper bei hohen Temperaturen überein. Bei niedrigen Temperaturen - und damit kann, je nach Substanz, durchaus auch Zimmertemperatur oder mehr gemeint sein - bricht die klassische Beschreibung zusammen, und wir müssen quantenmechanische Regeln anwenden, um
und andere thermodynamische Observable richtig vorhersagen zu können (siehe Kapitel 5).
translatorischen und
rotatorischen Freiheitsgraden hat die Hamiltonfunktion
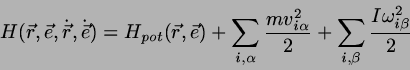
die Richtungsvektoren der linearen Moleküle,
deren Winkelgeschwindigkeit und
das molekulare Trägheitsmoment bezeichnet. Somit ist
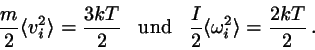
![\begin{displaymath}
\mu \equiv \frac{d A}{dN} = -kT \frac{d\ln{Q}}{dN}
= -kT \l...
... m k T}{h^{2}} \right)^{3/2}
\right] = - 6.08 \cdot 10^{-20} J
\end{displaymath}](img852.png)
![\begin{displaymath}
\langle a \rangle_{N,V,T} = \int a(\vec{\Gamma })
\exp{[-E(...
...\vec{\Gamma}
/ \int \exp{[-E(\vec{\Gamma })/kT]}d\vec{\Gamma }
\end{displaymath}](img856.png)
![\begin{displaymath}
\langle a \rangle_{N,V,T} = \int a(\vec{\Gamma}_{c})
\exp{[...
.../ \int
\exp{[-E_{pot}(\vec{\Gamma}_{c})/kT]}d\vec{\Gamma}_{c}
\end{displaymath}](img859.png)
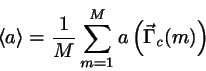
![\begin{figure}\includegraphics[width=300pt]{fig/f4kg.ps}
\end{figure}](img795.gif)