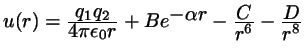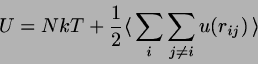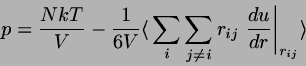Hard dumbbells,
hard spherocylinders, etc. |
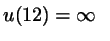 if overlap if overlap
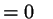 otherwise otherwise
|
First approximation to rigid molecules
|
|
Interaction site models, rigid |
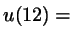 sum of isotropic pair energies
sum of isotropic pair energies
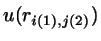 ,
where several
interaction sites ,
where several
interaction sites 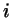 and and 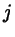 are in fixed positions on molecules are in fixed positions on molecules
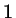 and and 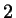 , respectively , respectively
|
Rigid molecules
|
|
Interaction site models with non-rigid bonds |
sum of isotropic pair energies, both intra- and intermolecular
|
Non-rigid molecules
|
|
Kramers-type |
sum of isotropic pair energies, exclusively between sites on different
molecules; certain intramolecular distances (bonds) and/or angles
are fixed
|
Flexible molecules, from ethane to biopolymers
|
|
Stockmayer |
Lennard-Jones + point dipoles
|
First approximation to small polar molecules |
|
Anisotropic Kihara |
![$u(12)=
4 \epsilon \left[ \left(
\frac{\textstyle{\rho_{12}}}{\textstyle{\si...
...
- \left(
\frac{\textstyle{\rho_{12}}}{\textstyle{\sigma}}\right)^{-6} \right] $](img26.png)
where 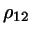 is the shortest distance between two linear rods is the shortest distance between two linear rods
|
Rigid linear molecules
with distributed
Lennard-Jones interaction |
|
Gay-Berne |
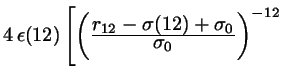
![$ - \left. \left(
\frac{\textstyle{r_{12}-\sigma(12)+\sigma_{0}}}{\textstyle{\sigma_{0}}}\right)^{-6} \right]
$](img29.png)
where 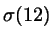 and and 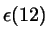 depend on depend on
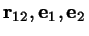 and certain substance-specific
shape parameters and certain substance-specific
shape parameters
|
Liquid crystal molecules of ellipsoidal shape, with
smoothly distributed Lennard-Jones sites
|
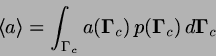
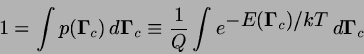
![\begin{displaymath}
u(r)= 4 \epsilon \left[ \left(\frac{r}{\sigma}\right)^{-12}
- \left(\frac{r}{\sigma}\right)^{-6} \right]
\end{displaymath}](img11.png)
![$\textstyle{
4 \epsilon \left[ \left(
\frac{\textstyle{r-a}}{\textstyle{\sigm...
...12}
- \left(\frac{\textstyle{r-a}}{\textstyle{\sigma-a}}\right)^{-6} \right]
}
$](img13.png)
![$u(r)=
A \left[ e^{\textstyle{-2b(r\!-\!r_{0})}}-
2 e^{\textstyle{-b(r\!-\!r_{0})}} \right] $](img17.png)