On an Ising lattice, draw spin values with equal probabilities
for and .
Molecules in disordered media:
Overlaps must be avoided.
Place the molecules on a lattice, then ``melt''this crystal before
the actual simulation run: Thermalization.
Population number in a cubic cell with face-centered cubic arrangement:
, with . Therefore typical particle numbers in
simulations are
etc.
What about 2-dimensional systems?
When simulating two-dimensional systems, the setting up of periodic
base cells and of initial configurations is an issue that takes some
considering. Since 2D systems are important both in the teaching and
in the application of simulation, some suggestions are compiled here.
A dense packing of discs is hexagonal. The most convenient periodic
cell in two dimensions would be quadratic. Unfortunately, these are
contradictory requirements.
There are two possible periodic cells compatible with a hexagonal
structure: (a) rectangular; (b) rhombic.
Rectangular unit cell:
Figure 2.2:
2D periodic cell: rectangle
The unit cell then has
and contains
discs.
Periodic boundary conditions are handled as usually, but with different
base cell lengths along the axes:
,
( may or may not be equal to .)
Nearest image convention: as usual, but again with different thresholds
along the axes: and , respectively.
Rhombic unit cell:
Figure 2.3:
2D periodic cell: rhombic
The crystallographic unit is now a rhombus with and
; it contains particle.
Periodic boundary conditions are best handled in a non-orthogonal
coordinate system. Let
denote the cartesian
coordinates, and
the coordinates in the
rhombic system. Then
with
(2.1)
Specifically, when the new coordinates (after a time or MC step) are
, then
Compute
(2.2)
(2.3)
Apply PBC as always, but in rhombic coordinates:
(2.4)
(2.5)
Now transform back to Cartesius:
(2.6)
(2.7)
Nearest image convention:
Is applied in cartesian coordinates, possibly with a potential cutoff
at :
(2.8)
(2.9)
Reference Density in 2D: Let be a reference distance, which
in the Lennard-Jones case is equal to
, and for hard discs is
identical to the disc diameter. The reference density is then
(2.10)
Let be a desired reduced density,
.
Thus, must be chosen as
.
Adjusting density and temperature:
Given , the density is adjusted to a desired value by shrinking or expanding
the volume: scale all coordinates by a suitable factor.
The temperature is a constant parameter in an MC simulation.
In molecular dynamics it must be adjusted in the following manner.
Since
(with
) or, in reduced units,
we first take the average of
over a
number of MD steps to determine the actual temperature of the simulated
system. Then we scale each velocity component according to
Since is a fluctuating quantity it can be adjusted
only approximately.
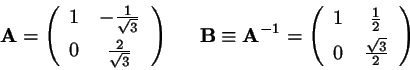
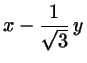
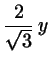
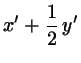
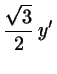
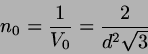
![\begin{figure}\includegraphics[width=330pt]{figures/rectcell.ps}
\end{figure}](img89.png)
![\begin{figure}\includegraphics[width=330pt]{figures/rhomcell.ps}
\end{figure}](img98.png)